
Mi profesor de cirugía, el recordado Dr. Luis Carrillo Maurtua, en una de sus clases magistrales sobre pancreatitis aguda, cuando describía de una manera tan estremecedoramente vívida el intenso e inconfundible dolor abdominal de esta enfermedad nos dijo al final, “sin embargo, la mayoría de los casos de pancreatitis aguda que van a ver, no serán así de típicos”. Y tenía razón, en la vida real, después de más de tres décadas de ejercer la medicina, uno termina convencido que para el diagnóstico de la mayoría de enfermedades relevantes hay que hacer un auténtico esfuerzo de observación, acopio de datos y reflexión ya que lo típico, como está descrito en los libros de medicina, ocurre más bien por excepción. Quizás esta sea la característica más fascinante de esta profesión. Por algo William Osler decía que “la medicina es el arte de la incertidumbre y la ciencia de la probabilidad”.
 La Fibrosis Quística no es una excepción. La forma típica de presentación de esta enfermedad, la de un niño de unos 2 años de edad con infección respiratoria (bronquitis, neumonía o sinusitis) persistente o recurrente, con deterioro progresivo de la función respiratoria, heces voluminosas, grasosas, de olor rancio y desnutrición, a pesar de buen apetito, se da pero pocas veces con esta combinación tan lógica y obvia de síntomas y signos. Después de todo, la Fibrosis Quística es una enfermedad que resulta de una mutación genética del gen que codifica la síntesis de una proteína llamada CFTR (Cystic Fibrosis Transmembrane Conductance Regulator – regulador de la conductancia transmembrana de la fibrosis quística), localizado en el brazo largo del cromosoma 7. Las posibles mutaciones de este gen son numerosas (se estima actualmente en cerca de 2,000 diferentes mutaciones). La expresión fenotípica (los síntomas de presentación) de estas mutaciones ocurre, como es predecible, en múltiples combinaciones.
La Fibrosis Quística no es una excepción. La forma típica de presentación de esta enfermedad, la de un niño de unos 2 años de edad con infección respiratoria (bronquitis, neumonía o sinusitis) persistente o recurrente, con deterioro progresivo de la función respiratoria, heces voluminosas, grasosas, de olor rancio y desnutrición, a pesar de buen apetito, se da pero pocas veces con esta combinación tan lógica y obvia de síntomas y signos. Después de todo, la Fibrosis Quística es una enfermedad que resulta de una mutación genética del gen que codifica la síntesis de una proteína llamada CFTR (Cystic Fibrosis Transmembrane Conductance Regulator – regulador de la conductancia transmembrana de la fibrosis quística), localizado en el brazo largo del cromosoma 7. Las posibles mutaciones de este gen son numerosas (se estima actualmente en cerca de 2,000 diferentes mutaciones). La expresión fenotípica (los síntomas de presentación) de estas mutaciones ocurre, como es predecible, en múltiples combinaciones.
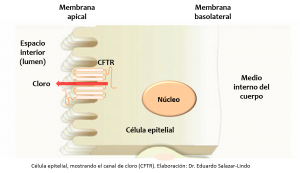 La proteína CFTR reside en la membrana apical de las células epiteliales que recubren las estructuras tubulares distribuidas por todo el cuerpo (por simplicidad, aquí las vamos a llamar ductos). CFTR funciona como un canal que facilita la salida de cloro y agua de la célula epitelial hacia el lumen (el espacio interior del ducto). Esta función es muy importante para humectar y mantener fluidas las secreciones que llegan al lumen de los ductos. El defecto básico de la Fibrosis Quística es la ausencia o mal funcionamiento de la proteína CFTR lo que ocasiona una incapacidad de mantener desatascados los ductos distribuidos por varias partes del cuerpo Por ejemplo, los bronquios y los ductos que intercomunican los senos paranales (aquellos que se enferman cuando hay sinusitis) están tapizados por una fina membrana epitelial que secreta una sustancia viscosa (moco) que normalmente está bien humectada y fluye libremente al exterior. En la Fibrosis Quística, este moco es extremadamente viscoso, seco, no fluye, se queda adherido al ducto, se acumula y lo obstruye. Esto explica los síntomas respiratorios recurrentes y las complicaciones infecciosas que padecen estos niños. Otro tanto ocurre en los ductos del páncreas que se atascan e impiden la salida de las enzimas digestivas, dificultando la digestión de los alimentos y originando el síndrome de malabsorción (principalmente de grasas) con pobre ganancia de peso y desnutrición.
La proteína CFTR reside en la membrana apical de las células epiteliales que recubren las estructuras tubulares distribuidas por todo el cuerpo (por simplicidad, aquí las vamos a llamar ductos). CFTR funciona como un canal que facilita la salida de cloro y agua de la célula epitelial hacia el lumen (el espacio interior del ducto). Esta función es muy importante para humectar y mantener fluidas las secreciones que llegan al lumen de los ductos. El defecto básico de la Fibrosis Quística es la ausencia o mal funcionamiento de la proteína CFTR lo que ocasiona una incapacidad de mantener desatascados los ductos distribuidos por varias partes del cuerpo Por ejemplo, los bronquios y los ductos que intercomunican los senos paranales (aquellos que se enferman cuando hay sinusitis) están tapizados por una fina membrana epitelial que secreta una sustancia viscosa (moco) que normalmente está bien humectada y fluye libremente al exterior. En la Fibrosis Quística, este moco es extremadamente viscoso, seco, no fluye, se queda adherido al ducto, se acumula y lo obstruye. Esto explica los síntomas respiratorios recurrentes y las complicaciones infecciosas que padecen estos niños. Otro tanto ocurre en los ductos del páncreas que se atascan e impiden la salida de las enzimas digestivas, dificultando la digestión de los alimentos y originando el síndrome de malabsorción (principalmente de grasas) con pobre ganancia de peso y desnutrición.
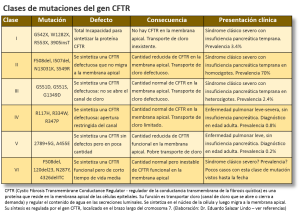 La Fibrosis Quística involucra diversos grados y mecanismos de mal funcionamiento de las células epiteliales del intestino, sistema respiratorio, páncreas, hígado y vías biliares, sistema reproductivo y glándula sudoríparas. La severidad de la obstrucción de los ductos, la edad de presentación, el grado de compromiso de los diferentes sistemas ductales, varía según la mutación que está en juego. A la fecha se reconocen seis clases de mutaciones según cuál sea el defecto básico en la síntesis del CFTR. La tabla muestra las mutaciones más comunes, el tipo de defecto, las consecuencias y la forma de presentación clínica de cada una de estas seis clases. Las clases I, II y III son las formas más clásicas y más conocidas. Las clases IV, V y VI corresponden a las formas menos típicas y que pueden presentarse recién en la vida adulta. Se estima que un buen porcentaje de personas con estas formas atípicas de Fibrosis Quística viven sin saber que tienen esta enfermedad. Los médicos debemos aumentar nuestro grado de alerta acerca de la Fibrosis Quística, no solo porque el diagnóstico más precoz mejora el pronóstico considerablemente, sino porque hay formas clínicas menos severas pero trascendentes que pasan sin diagnóstico.
La Fibrosis Quística involucra diversos grados y mecanismos de mal funcionamiento de las células epiteliales del intestino, sistema respiratorio, páncreas, hígado y vías biliares, sistema reproductivo y glándula sudoríparas. La severidad de la obstrucción de los ductos, la edad de presentación, el grado de compromiso de los diferentes sistemas ductales, varía según la mutación que está en juego. A la fecha se reconocen seis clases de mutaciones según cuál sea el defecto básico en la síntesis del CFTR. La tabla muestra las mutaciones más comunes, el tipo de defecto, las consecuencias y la forma de presentación clínica de cada una de estas seis clases. Las clases I, II y III son las formas más clásicas y más conocidas. Las clases IV, V y VI corresponden a las formas menos típicas y que pueden presentarse recién en la vida adulta. Se estima que un buen porcentaje de personas con estas formas atípicas de Fibrosis Quística viven sin saber que tienen esta enfermedad. Los médicos debemos aumentar nuestro grado de alerta acerca de la Fibrosis Quística, no solo porque el diagnóstico más precoz mejora el pronóstico considerablemente, sino porque hay formas clínicas menos severas pero trascendentes que pasan sin diagnóstico.
Referencias
- Orenstein DM, Rosenstein BJ, Stern RC, eds. Cystic Fibrosis Medical Care. Philadelphia: Lippincott Williams & wilkins, 2000
- CFTR.Info. CFTR Mutations. Understanding how CFTR mutations influence disease prognosis. http://www.cftr.info/about-cf/cftr-mutations/
- Petitt RS. Cystic Fibrosis Transmembrane Conductance Regulator–Modifying Medications. http://www.medscape.com/viewarticle/768501_2